|
|
Our research has been based on
the first principle methods for computation the
electronic structure and electron transport of
mesoscopic system that enables modelling of the
system independently of all parameters related to
the system.
We have focused on applying these methods to
nanostructures such as carbon nanotubes and
graphene.
The computation of electronic transport properties
of these systems has been performed by using
non-equilibrium Green's function (NEGF) and density
functional theory (DFT) with the help of software
packages such as Siesta, TranSiesta and TBTrans.
This simulation tool has been used to gain insight
into the performance and evolution of these systems,
and as an attempt to sequence the base within DNA
molecule positioned between two nanotubes and in
pore in the graphene by measuring the electrical
currents
through
the system at a given voltage.
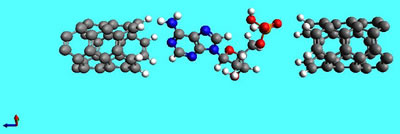
FIG.1. Adenine, base of DNA, between two armchair
(3,3) nanotubes
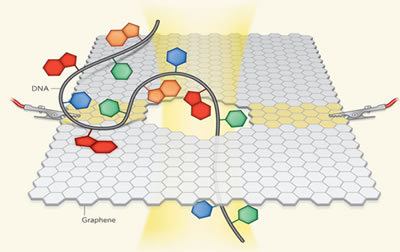
FIG.2.Single-stranded
DNA translocate through the graphene |